KRAS mutations have wide-spread prevalence in human cancers nowadays. The mis-sense mutation of KRAS at codon 12 aberrantly activates the protein into a hyperexcitable state. Attenuating its GTPase activity results in accretion of GTP-bound activated KRAS and activation of downstream signaling pathways. Ideally, the KRASG12C protein product is an attractive, yet challenging, target for small molecule inhibition. Crucially, KRASG12C is a mutant form of the guanosine triphosphatase (GTPase) KRAS. Collectively, inhibitors targeting KRASG12C are a promising new class of oncogene-specific therapeutics for the treatment of tumors driven by the mutant protein. Besides, these KRASG12C inhibitors react with the mutant cysteine residue. The effect by binding covalently to the switch-II pocket (S-IIP) that is present only in the inactive guanosine diphosphate (GDP)–bound form of KRASG12C, sparing the wild-type protein. Fortunately, researchers found that ARS-1620 was an potent KRASG12C inhibitor.
In this study, ARS-1620 induces tumor regression through an on-target mechanism of action. Especially, ARS-1620 is an atropisomeric selective KRASG12C inhibitor with desirable pharmacokinetic (PK). Particularly, ARS-1620 selectively induces tumor regression in patient-derived tumor models. Moreover, ARS-1620 is a valuable pharmacological tool to interrogate KRAS biology in vivo. In addition, ARS-1620 exhibits excellent oral bioavailability in mice and sufficient blood stability essential for quantitative measurements of in vivo G12C target occupancy. ARS-1620 displays potent and selective anti-tumor activity in patient-derived (PDX) tumor models. As a result, the in vivo evidence that ARS-1620 is broadly efficacious as a single agent across non-small-cell lung carcinoma (NSCLC) models.
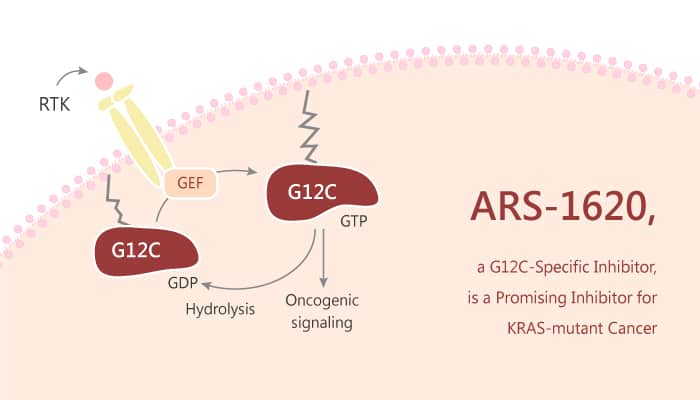
All in all, these results indicate that ARS-1620 represents a novel class of direct KRAS inhibitors with in vitro to in vivo potency and therapeutic window in the range of a drug candidate.