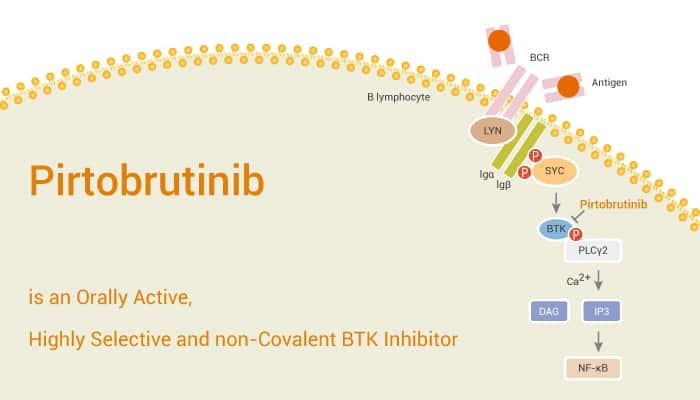
Bruton’s tyrosine kinase (BTK) is a non-receptor kinase that plays a crucial role in oncogenic signaling. It is critical for the proliferation and survival of leukemic cells in many B cell malignancies. BTK has emerged as a promising drug target for multiple diseases, particularly haematopoietic malignancies and autoimmune diseases related to B lymphocytes. Covalent BTK inhibitors have used in B-cell malignancies, but there they have off-target toxicity and resistance, leading to eventual treatment discontinuation and disease progression. In this study, Pirtobrutinib is a highly selective, non-covalent, next-generation BTK inhibitor.
Pirtobrutinib potently inhibits both wild-type (WT) BTK and BTK C481S -mediated kinase activity in enzyme and cell-based assays with nanomolar potency. It inhibits WT BTK (Y223) autophosphorylation with an IC50 of 3.68 nM. Pirtobrutinib inhibits BTK C481S Y223, C481T Y223, and C481R Y223 autophosphorylation. The IC50s are 8.45, 7.23, and 11.73 nM, respectively. Pirtobrutinib also causes regression of BTK-dependent lymphoma mouse xenograft models. Furthermore, it is more than 300-fold selective for BTK over 98% of 370 other kinases. Pirtobrutinib also shows no significant inhibition of non-kinase off targets at 1 mM. In addition, ADME and pharmacokinetic experiments in two preclinical species predict that Pirtobrutinib will have high human exposure and sustained BTK C481S target coverage at clinically achievable doses.
In summary, Pirtobrutinib is a next-generation, non-covalent, highly selective BTK inhibitor. It potently inhibits the cellular activity of BTK C481S, T, and R mutations. It also displays strong equilibrium binding to WT BTK and several BTK C481 substitution mutations. Pirtobrutinib may overcome acquired resistance to covalent BTK inhibitors in patients without significant off-target toxicity.
Reference:
Gomez E B, et al.. Blood, 2019, 134(Supplement_1):4644-4644.