Non-small cell lung cancers with activating mutations in the kinase domain of the EGFR demonstrate dramatically, but transient, responses to the reversible tyrosine kinase inhibitors Gefitinib. To identify mechanisms of resistance to Gefitinib, Researchers used NSCLC cells harboring an activating EGFR mutation to generate multiple resistant clones. The bronchoalveolar cancer cell line NCI-H1650 has an in-frame deletion of the EGFR kinase (delE746-A750).
Researchers cultured the bronchoalveolar cancer cell line NCI-H1650 in 20 μM Gefitinib. This cell line exhibits 100-fold increased sensitivity to Gefitinib, compared with some NSCLC lines expressing wild-type EGFR. Whereas 20 μM gefitinib efficiently killed the vast majority of these cells, drug-resistant colonies are readily irrespective of mutagen treatment. However, these cells display persistent sensitivity to HKI-357. HKI-357 is a dual inhibitor of EGFR and ERBB2 with IC50s of 34 nM and 33 nM, respectively. In particular, HKI-357 is an irreversible inhibitor, most likely via a covalent bond with the cys773 residue within the EGFR catalytic domain or the cys805 of ERBB2. Like Gefitinib, HKI-357 demonstrates increased killing of NSCLC cells harboring an EGFR mutation, compared with cells expressing the wild-type receptor.
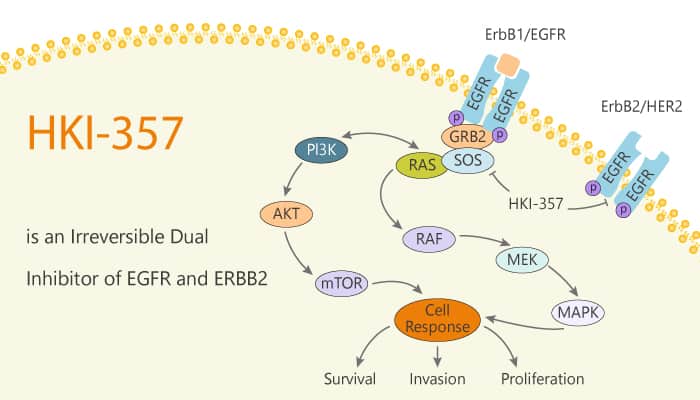
Inhibition of EGFR by an irreversible inhibitor is sufficient to induce apoptosis in Gefitinib-resistant cells. Treatment with HKI-357 inhibits EGFR autophosphorylation (Y1068). HKI-357 also inhibits phosphorylation of downstream effectors AKT and MAPK (ERK) in cells. Remarkably, HKI-357 is 10-fold more effective than Gefitinib in suppressing EGFR autophosphorylation, and AKT and MAPK phosphorylation in parental NCI-H1650 cells harboring the delE746-A750 EGFR mutation.
All in all, HKI-357 may prove highly effective in the treatment of EGFR-mutant NSCLCs, including tumors that have become resistant to Gefitinib.