JAK2 and STAT3 have been extensively implicated in the development and progression of numerous cancers. STAT3 be also activated by EGFR, JAK2, and other TYKs activated by EGF, LIF, and other cytokines. STAT3 constitutively activats in 60% of primary malignant gliomas, and the extent of activation correlates with glioma grade.
Gliomas are the most common primary brain tumors. Both EGFR and EGFRvIII strongly promote gliomagenesis and are promising potential therapy targets. Mechanisms underlying GBM resistance to anti-EGFR therapy are not entirely clear. However, phosphatase and tensin homolog (PTEN) deficiency and deregulated phosphatidylinositol 3-kinase (PI3K) pathway activity may play an important role because they correlate with resistance to EGFR inhibitors.
A study from Kunyan He, et al. has discovered a small-molecule inhibitor, G5-7, that selectively blocked JAK2-mediated phosphorylation of EGFR on Tyr1068 by allosterically binding JAK2, and was more potent in suppressing the proliferation of U87MG/EGFRvIII cells than were canonical EGFR and JAK2 inhibitors. Furthermore, G5-7 reduced vascular endothelial growth factor (VEGF) secretion and angiogenesis in GBM.
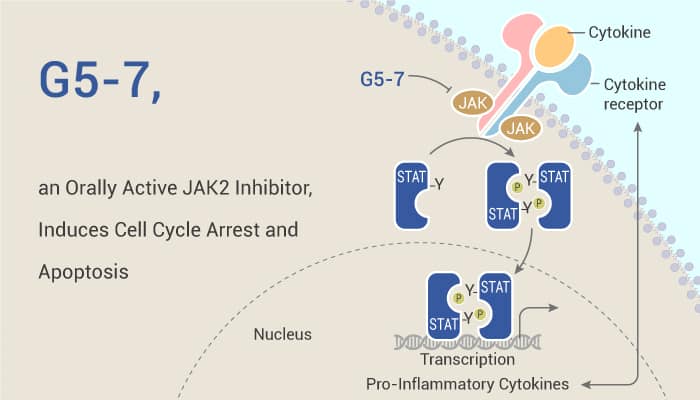
In vitro, G5-7 (0-5 μM) inhibits EGFR tyrosine phosphorylation and downstream mTOR signaling and arrests the cell cycle at the G2 phase. Also, it does not directly inhibit EGFR activation. Moreover, the compound (0-10 μM) comparably increases the abundance of markers (cleaved-PARP and caspase 3) of apoptosis in parental LN229 cells and U87MG/EGFRvIII cells. G5-7 interacts with full-length JAK2 and significantly inhibits EGFR Tyr1068 phosphorylation but had no effect on EGFR Tyr1045 phosphorylation. Especially, G5-7 downregulates the downstream signaling of JAK by mTOR.
In vivo, G5-7 (10 and 50 mg/kg, oral gavage, 5- to 6-week-old female nude mice) decreases VEGF secretion and exerts a potent antiangiogenic effect.
Reference: