Cancers arise owing to the accumulation of mutations in critical genes that alter normal programmes of cell proliferation, differentiation and death. The RAS–RAF–MEK–ERK–MAP kinase pathway mediates cellular responses to growth signals. Three RAF genes code for cytoplasmic serine/threonine kinases. Whereas mutations in ARAF and CRAF are very rare, BRAF mutations widely vary across multiple cancer types. BRAF is a serine/threonine kinase that is commonly activated by somatic point mutation in human cancer. Among a variety of BRAF alterations identified to date, point mutations affecting amino acid position 600 are by far the most frequent ones (V600E or V600K; melanoma, >90%). Subsequently, researchers initiate multiple drug discovery programs to synthesize potent and drug-like BRAF inhibitors.
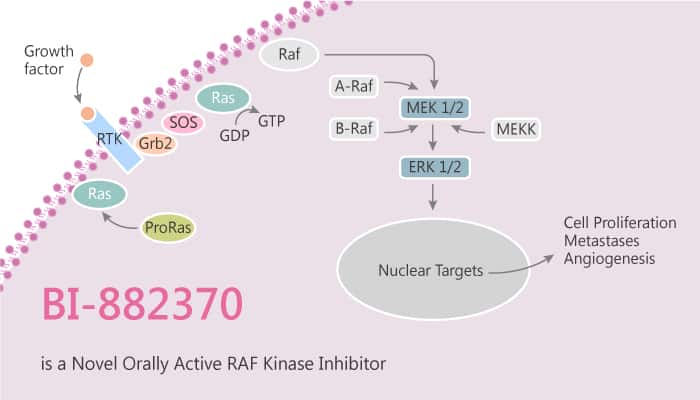
BI-882370 is a highly potent and selective RAF inhibitor that binds to the DFG-out (inactive) conformation of the BRAF kinase. Especially, BI-882370 potently inhibits the oncogenic BRAFV600E-mutant, WT BRAF and CRAF kinases with similar IC50s of 0.4, 0.8, and 0.6 nM, respectively). In particular, BI-882370 shows EC50s of 0.5 and 0.7 nM in A375 and SK-MEL-28 (BRAFV600E) melanoma cells, respectively. Furthermore, BI-882370 inhibits proliferation of human BRAF-mutant melanoma cells with 100× higher potency (1-10 nM) than Vemurafenib. However, BI-882370 does not affect wild-type cells at 1,000 nM.
BI-882370 is efficacious in multiple mouse models of BRAF-mutant melanomas and colorectal carcinomas. BI-882370 shows superior efficacy compared with Vemurafenib, Dabrafenib, or Trametinib. Moreover, BI-882370 induces phosphorylation of MEK1/2 and enhances phosphorylation of ERK1/2 in BRO cells at concentrations between 3 and 300 nM.
All in all, BI-882370 is a RAF inhibitor that exhibit a superior therapeutic window.