DNA-damaging chemotherapeutics, such as Cisplatin, have been the mainstay of cancer treatment for decades. These therapies generate DNA lesions, and high-fidelity replicative DNA polymerases as the template cannot utilization. Thus, it blocks the progression of the replication fork, generates cytotoxicity, and ultimately causes cell death. However, cells employ specialized DNA polymerases to bypass the lesion site at the cost of replication fidelity in a process. It is known as translesion synthesis (TLS). In addition, TLS contributes to chemoresistance as well as treatment-induced mutations, targeting TLS is an attractive avenue for improving chemotherapeutics. JH-RE-06 disrupts mutagenic TLS by preventing recruitment of mutagenic POL ζ.
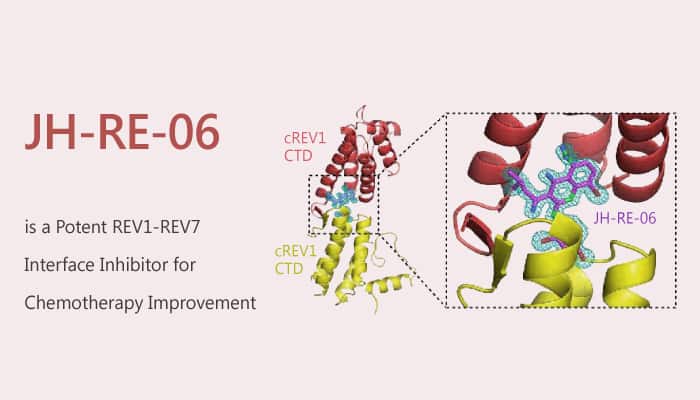
JH-RE-06, a potent REV1-REV7 interface inhibitor (IC50=0.78 μM; Kd=0.42 μM), targets REV1 that interacts with the REV7 subunit of POLζ. Furthermore, JH-RE-06 induces REV1 dimerization to block the REV1- REV7 interaction. JH-RE-06 acts by specifically inhibiting REV1-dependent mutagenic TLS. Thus, it has the potential to be a novel class of adjuvant which can sensitize cells to the cytotoxic effect of a DNA-damaging chemotherapeutic drug. Simultaneously, it suppresses the attendant mutagenesis caused by the treatment.
In vivo, JH-RE-06 or Cisplatin-alone treatments suggest that suppression of the REV1-dependent mutagenic TLS by JH-RE-06-mediated specific inhibition of the REV1-REV7 interaction significantly improves chemotherapy. Furthermore, Co-administration of JH-RE-06 with Cisplatin suppresses the growth of xenograft human melanomas in mice, establishing a framework for developing TLS inhibitors as a novel class of chemotherapy adjuvants.
In summary, as a potent REV1-REV7 interface inhibitor, JH-RE-06-induced asymmetric dimerization of the REV1 CTD at its conformationally flexible and shallow REV7-binding surface may furnish a new paradigm.
Reference:
Wojtaszek JL, et al. 2019 Jun 27;178(1):152-159.e11.